
A Sample of Current Projects
By Joseph Hargitai and Shenglong Wang
NYU’s high performance computing systems are a vital resource for University research in the sciences and across the disciplinary spectrum. A related article in this issue of Connect provides a description of the NYU High Performance Computing Service (NYU HPC), and a new support wiki that has been designed to make using HPC resources easier. Here is just a sampling of recent research performed using NYU HPC resources.
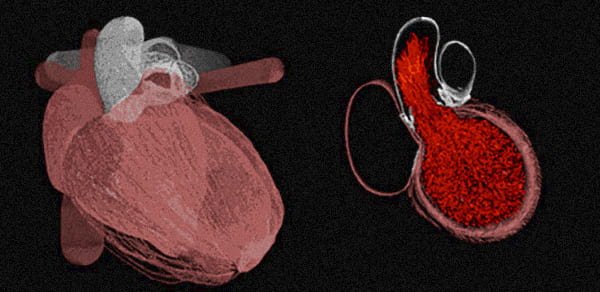
Modeling the Human Heart
Professor Charles Peskin (Mathematics, CIMS) and NYU researchers Professor Boyce Griffith (Division of Cardiology, NYU Langone Medical Center) and Dr. David McQueen (CIMS) have been developing computational models of several different aspects of cardiac physiology and pathophysiology. They are currently working to complete a model of cardiac fluid-structure interaction in the context of congestive heart failure. The anatomy of this model is derived from computed tomography imaging of a human patient, and its physiology is being tuned to correspond to that of the patient.
These researchers are also performing simulation-based studies of the fluid dynamics of the aortic heart valve. These simulations constitute some of the highest-resolution three-dimensional simulations of heart valve dynamics performed to date. A novelty in both studies is that these simulations are performed for multiple cardiac cycles, whereas earlier models were limited to only a single beat. Professor Griffith is also working with Professor Glenn I. Fishman, director of the Division of Cardiology at the NYU Langone Medical Center, and Dr. Paul Hand, a postdoctoral fellow at NYU, to study normal and abnormal electrical impulse propagation in cellular and multiscale models of cardiac muscle fibers, as well as in tissue-level descriptions of the heart.
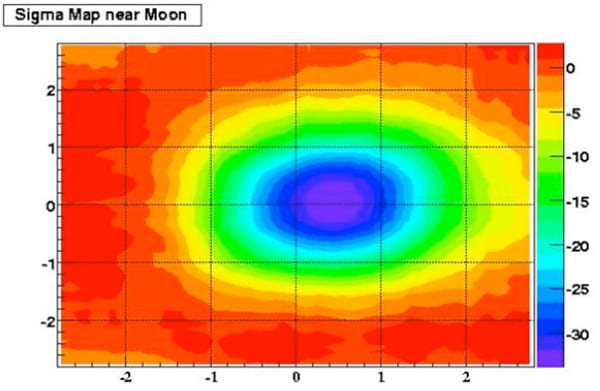
Learning about the Sun and High-Energy Cosmic Rays
Professor Allen Mincer (Physics, FAS) and his Ph.D. student Grant Christopher have been performing a series of simulations to increase the understanding of data obtained from the MILAGRO (Multiple Institution Los Alamos Gamma Ray Observatory) detector in the Jemez Mountains near Los Alamos, New Mexico.
Professor Mincer writes “Cosmic rays coming from the direction of the sun or moon (but originating much further away) may hit the sun or moon and not make it to Earth. This can be detected here on Earth as a paucity of cosmic rays coming from these directions, effectively a solar or lunar cosmic-ray shadow.
“Particle air-showers from TeV (very high energy) cosmic rays have been measured using MILAGRO. Since we know the Earth’s magnetic field, measuring the lunar shadow with MILAGRO tells us about the detector’s angular resolution and allows calibrating the detector’s energy scale. Measurement of the solar shadow can be used to obtain information about the interplanetary solar magnetic field and therefore teach us about the Sun. Looking for a second lunar shadow deflected by the Earth’s field in the direction opposite the main shadow allows setting limits on the antiparticle content of cosmic rays.”
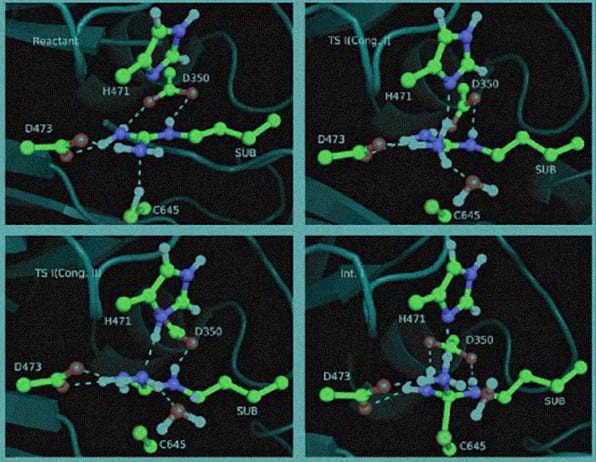
Computer Simulations of Enzyme Reactions
Professor Yingkai Zhang (Chemistry, FAS) and his research group have been using the NYU HPC resources in computational studies of enzyme reactions.
Professor Zhang writes, “Enzymes are remarkable catalysts provided by nature, and play essential roles in cells of all organisms. Fully characterizing their complicated reaction mechanisms is a fundamental goal and challenge in biological chemistry, and has important implications in rational drug design and the discovery of novel biocatalysts.
“In computational studies of enzyme reactions, extensive sampling on a reasonably accurate potential energy surface is needed to obtain reliable results. In the last few years, my research group has advanced the frontier of a much-desired approach, called Born-Oppenheimer ab initio QM/MM molecular dynamics simulations, to simulating enzyme reactions. Such simulations until now have been widely considered to be computationally too expensive to be feasible. Using the HPC resources provided by NYU ITS, Professor Zhang’s group has demonstrated this state-of-the-art approach to be viable and successful in investigating several medically important and biologically significant enzymes.”
Co-Regulated Gene Modules: Similarities across Species
Professor Richard Bonneau (Biology, FAS) and his research group have been using NYU HPC resources, and a novel method of analysis, to uncover multi-species similarities in gene regulation. Peter Waltman is a doctoral student working within Professor Bonneau’s group. He writes, “A key challenge in the analysis of genomics data is the identification of modules of genes with similar or identical regulatory controls, a nontrivial problem due to the complexity of regulatory networks. The results from several recent works suggested that many co-regulated modules are conserved (in whole or in part) across several species, and that an integrative, comparative analysis of multiple functional genomics data types could prove powerful in accurately identifying those co-regulated groups of modules that are conserved across multiple species for which genomics data collections exist.
“Using the high performance computing clusters provided and maintained by the NYU HPC, we performed several multi-species analyses using an extension of our cMonkey algorithm that allows for the simultaneous biclustering of heterogeneous data compendia spanning multiple prokaryotic species. The analyses were performed on a group of Gram positive species (containing Bacillus subtilis, Bacillus anthracis, and Listeria monocytogenes) and a group of Gram negative species (containing Escherichia coli, Salmonella typhimurium and Vibrio cholerae).
“These analyses identified conserved modules of orthologous genes, yielding evolutionary insights into the formation and surprisingly high degree of conservation of regulatory modules. We also identified similarities and differences over the experimental conditions under which the conserved modules are active, such as a temporal difference between the two Bacillus species in the expression of genes required for spore formation.
The NYU HPC also proved invaluable to the performance of numerous, high-throughput, statistical validations of our novel method, which we are currently in the process of publishing with a focus on the results from the multi-species biclustering of the Gram positive group.”
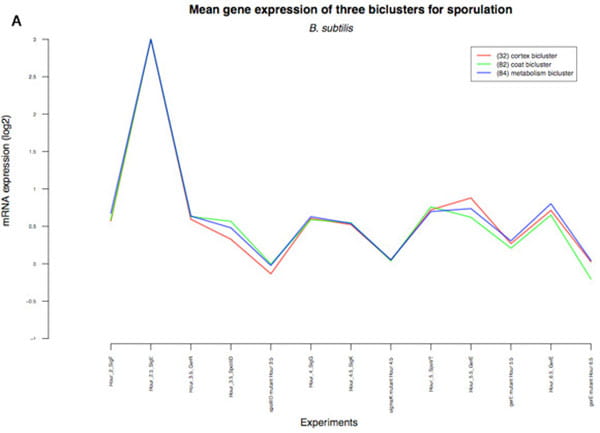
The figures below (see photo gallery) shows expression profiles of three partially conserved sporulation biclusters, identified by the multi-species analysis of B. subtilis and B. anthracis. The B. subtilis biclusters all follow the same expression profile (i.e., similar expression over nearly every experimental condition included in the dataset), while the B. anthracis biclusters display distinct profiles, revealing a temporal aspect not present in the B. subtilis dataset. Bicluster 84 (blue line) is composed primarily of genes involved in metabolic functions during sporulation, bicluster 82 (green line) includes primarily genes encoding spore coat proteins, and 32 (red line) contains genes involved in spore cortex formation and activation of the σ factors required for the latest stages of sporulation.

(B) The expression profiles of the B. subtilis biclusters, for the bicluster conditions involved in sporulation. |

(C) The expression profiles of the B. subtilis biclusters, for all bicluster conditions including those not involved in sporulation. |
Monte Carlo Simulations of Chromatin Folding
Members of the biomolecular simulation group led by Professor Tamar Schlick within the FAS Department of Chemistry are currently using the NYU HPC clusters to investigate the effects of different internal and external factors on DNA compaction and folding by means of an innovative mesoscale model and Monte Carlo simulations.
Professor Schlick writes: “In eukaryotic cells, the chromosome material is organized in a protein/DNA complex called the chromatin fiber. To fit into the micron-sized cells, such fibers must rearrange to condense the genetic material by more than 5 orders of magnitude. Despite intense research by both modelers and experimentalists over the past few decades, the detailed organization, let alone its folding dynamics and energetics of the chromatin fiber, are not well understood.
“Understanding chromatin organization is of fundamental importance because the state of DNA affect DNA’s accessibility to the protein machinery, which in turn directly affects transcription regulation. A realistic computational model can provide molecular-level views and quantitative information that cannot be easily revealed from experimental studies to shed insights into this challenging problem.”
The team, including researchers Ognen Perisic, Rosana Collepardo, and Hin Hark Gan are currently using the HPC clusters extensively to investigate the effects of different internal and external factors on DNA compaction and folding by means of an innovative mesoscale model and Monte Carlo simulations for configurational sampling.
The mesoscale model was developed and refined by Professor Schlick’s research group over the past ten years and is capable of simulating chromatin dynamics and predicting chromatin’s internal organization. The model captures the essential physics of chromatin, such as its electrostatics, DNA and nucleosome mechanics, structural irregularity, and histone tail flexibility, and averages over other effects: protein/DNA sequence effects, hydrogen-bonding, atomistic fluctuations, and solvation effects. Recent studies have elucidated the effect of divalent ions on chromatin structure2-4 and the effect of variations in the nucleosome repeat length on chromatin geometry.5,6
The team’s long-term research goals are to integrate structural and dynamical aspects of chromatin organization to delineate the mechanisms of transcriptional regulation mediated through protein factors, epigenetic marks, and environmental conditions.

Monte Carlo Simulations of Chromatin Folding: The coarse-grained chromatin model. |

Monte Carlo Simulations of Chromatin Folding: Chromatin folding. |
Footnotes
- Image reprinted with permission from “Active Site Cysteine Is Protonated in the PAD4 Michaelis Complex: Evidence from Born-Oppenheimer Ab Initio QM/MM Molecular Dynamics Simulations,” by Z. Ke, Y. Zhou, P. Hu, S. Wang, D. Xie and Y. Zhang, J. Phys. Chem. B, 113, 12750-12758, (2009). Copyright 2009 American Chemical Society.
- S. Grigoryev, G. Arya, S. Correll, C. Woodcock, and T. Schlick, “Evidence for hetromorphic chromatin fibers from analysis of nucleosome interactions.”, Proc. Natl. Acad. Sci. USA, 106: 13317-13322 (2009).
- G. Arya and T. Schlick, “A tale of tails: how histone tails mediate chromatin compaction in different salt and linker histone environments” J. Phys. Chem. A, 16: 4045-4059 (2009).
- H. Gan, O. Perisic and T. Schlick, “The chromatin ionic atmosphere in monovalent and divalent salts analyzed by a mesoscale electrostatic model”, To be submitted (2010).
- T. Schlick and O. Perisic, “Mesoscale simulations of two nucleosome-repeat length oligonucleosomes.”, Phys. Chem. Chem. Phys., 11: 10729-10737 (2009).
- O. Perisic, R. Collepardo-Guevara and T. Schlick, “Modeling studies of chromatin fiber structure as a function of DNA linker length”, J. Mol. Biol. In Revision (2010).
About the Authors
Faculty Technology Specialist Joseph Hargitai and System Administrator Shenglong Wang worked within ITS’ High Performance Computing group at the time of this publication.